
2月12日,生物学领域重要期刊Briefings in Bioinformatics在线发表了我校人工智能学院计智伟教授课题组题为“ULDNA: Integrating Unsupervised Multi-Source Language Models with LSTM-Attention Network for High-Accuracy Protein-DNA Binding Site Prediction”的研究论文。针对蛋白质-DNA结合位点预测问题,他们开发了一种新的深度学习预测方法ULDNA。

ULDNA 的核心思想是利用蛋白质大语言模型针对序列设计特征表示,再结合注意力机制的长短期记忆网络(LSTM-Attention Network)训练DNA结合位点预测模型。研究人员选取了PDNA-128、PDNA-316、PDNA-335等7个基准数据集(蛋白质序列数目从40到600不等),对ULDNA进行了全面测试。实验结果表明,ULDNA在所有数据集上均表现卓越,预测性能明显优于其他9种主流方法,包括TargetDNA (IEEE/ACM Trans Comput Biol Bioinform, 2016)、DNAPred (J Chem Inf Model, 2019)、GraphBind (Nucleic Acids Res, 2021)、NCBRPred (Brief Bioinform, 2021)、PredDBR (IEEE/ACM Trans Comput Biol Bioinform, 2021)、iDRNA-ITF (Brief Bioinform, 2022)等。ULDNA的性能优势主要归因于两点创新:首先,三种不同的蛋白质大语言模型从海量蛋白质序列中学习了丰富的、互补的进化知识,能够将蛋白质序列编码为蕴含DNA结合模式的、高度可鉴别性的特征表示;其次,LSTM-Attention Network能够有效地挖掘进化知识和蛋白质-DNA结合模式之间的深层次映射机制。此项研究提供了一种新的高通量生物计算方法,它能够快速准确地从大规模蛋白质序列中预测出潜在的DNA结合位点。
研究人员首先在PDNA-128测试集上比较ULDNA和9种主流的蛋白质-DNA结合位点预测方法的性能,其中PDNA-128包含了128条在2023年1月以后加入蛋白质结构数据库PDB的蛋白质序列。从表1中可见,ULDNA的MCC (Mathew’s Correlation Coefficient)、AP (Average Precision)和AUROC (Area Under the Receiver Operating Characteristic Curve)值在所有方法中均排名第一。相比于排名第二的GraphSite,ULDNA在上述三个评价指标上分别增长了6.1%、5.8%和1.6%。

表1. ULDNA和9种蛋白质-DNA结合位点预测方法在PDNA-128测试集上的性能比较
进一步,研究人员分别单独或联合利用蛋白质大语言模型ESM2、ProtTrans和ESM-MSA transformer抽取序列的特征表示,再结合LSTM-Attention Network进行DNA结合位点预测,进而分析每种大语言模型对ULDNA的贡献度,如图1所示。在5组蛋白质-DNA结合位点数据集上的实验结果表明,ESM2+ProTrans+ESM-MSA组合模型的预测性能优于其他6个独立/组合模型,并且较之组合模型ProtTrans+ESM-MSA的提升最明显。上述实验结果证明了两点:首先,三种蛋白质大语言模型均有助于提升预测精度;其次,ESM2对ULDNA的贡献度最大。
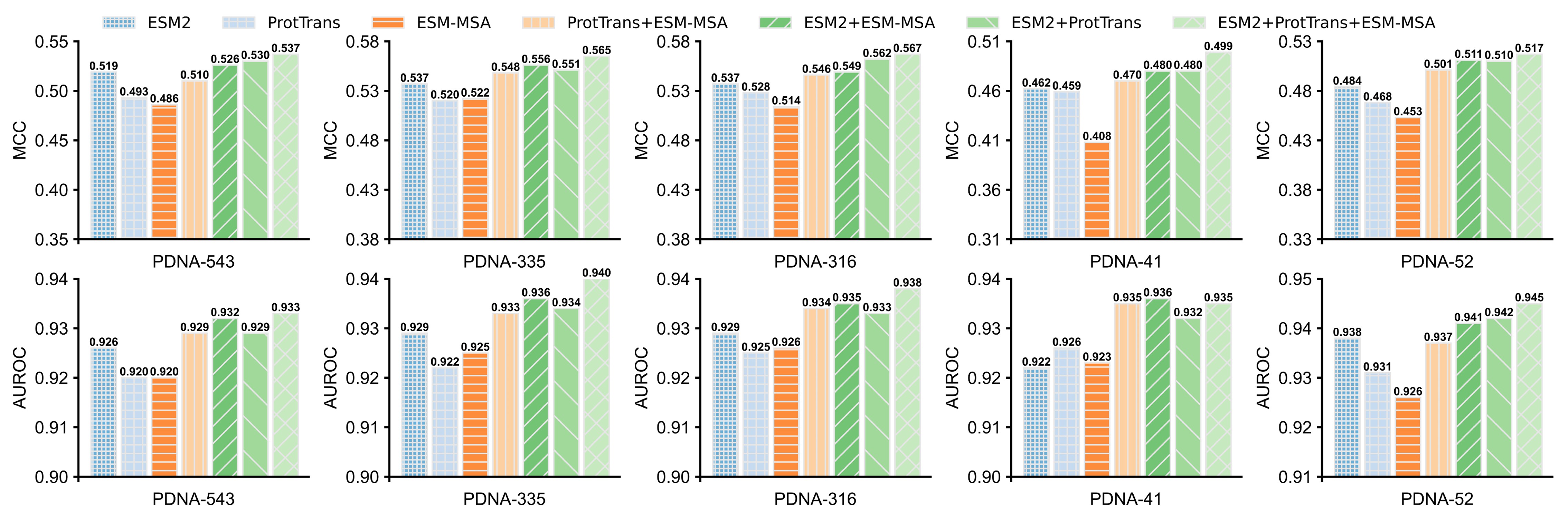
图1. 不同蛋白质大语言模型在蛋白质-DNA结合位点预测中的性能比较
此外,研究人员挑选了两个蛋白质2MXF_A and 3ZQL_A进行实例分析。在每个蛋白质上,比较四种内部方法(LA-ESM2、LA-ProtTrans、LA-ESM-MSA和ULDNA)和外部方法PredDBR的预测性能,并使用PyMOL软件将预测结果可视化,如图2所示。可以观察出两个有趣的现象:首先,ULDNA在每个蛋白质上拥有最多的真阳性位点和最少的假阳性位点,因此它的综合预测性能远优于其它4种对比方法;其次,三种内部方法LA-ESM2、LA-ProtTrans和LA-ESM-MSA的预测结果能够相互补充,进而导致融合后ULDNA的性能取得了进一步提升。
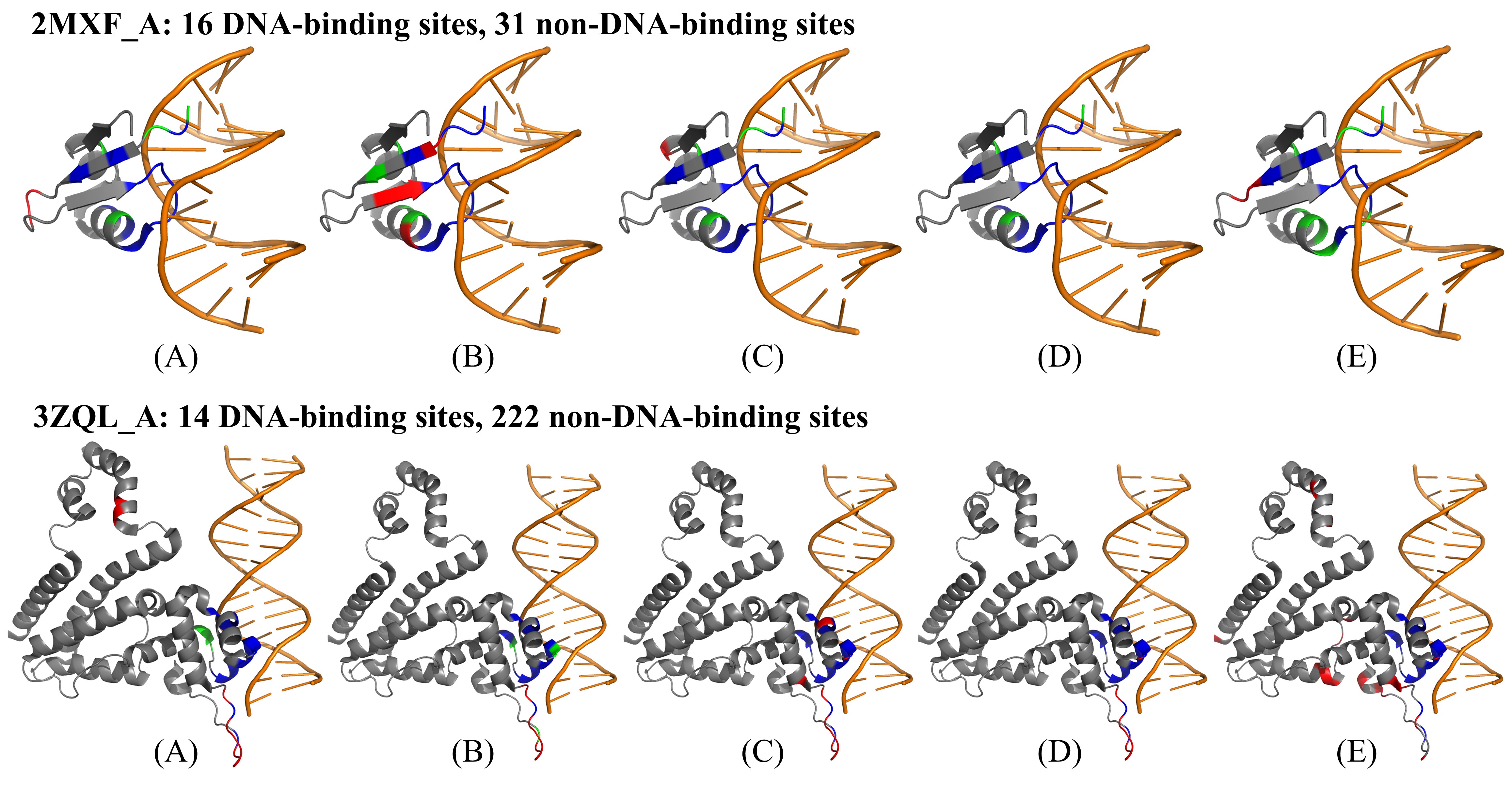
图2. ULDNA和4种预测方法在蛋白质2MXF_A和3ZQL_A上的可视化预测结果。(A) LA-ESM2, (B) LA-ProtTrans, (C) LA-ESM-MSA, (D) ULDNA, (E) PredDBR;橙色表示DNA分子,灰色表示蛋白质分子;红色、蓝色和绿色分别表示假阳性位点(非DNA结合位点被错误地预测为结合位点)、真阳性位点(被正确预测的DNA结合位点)和假阴性位点(DNA结合位点被错误地预测为非结合位点)。
综上所述,该研究提出了一种基于深度学习的蛋白质-DNA结合位点预测方法ULDNA,未来有望辅助靶向药物筛选。研究结果证实,ULDNA的预测性能要显著地优于主流方法。更重要的是,该研究证明了多种蛋白质大语言模型在蛋白质-DNA结合位点预测中的潜在有效性和互补性。然而,该研究也存在一些不足和挑战。例如:目前使用的线性特征融合策略容易造成信息冗余,该策略将在后续研究工作中被进一步地优化;再者,考虑到蛋白质结构预测方法(如AlphaFold和ESMFold)的快速发展,后续可能在蛋白质-DNA结合位点预测中融入预测的蛋白质结构信息,以期进一步提升预测精度。
本文的第一作者是我校人工智能学院讲师朱一亨博士,通讯作者是计智伟教授和南京理工大学计算机科学与工程学院於东军教授。感谢我校海外高层次引进人才启动项目、国家自然科学基金项目、江苏省自然科学基金项目、江苏省农业科技自主创新项目、中央高校基本业务经费等项目的支持。
原文链接:https://doi.org/10.1093/bib/bbae040