近日,南京农业大学免疫研究所以第一完成单位与扬州大学和中国科学院微生物研究所等单位,在病毒学领域国际顶级期刊《Trends in Microbiology》上发表论文“Epidemiology, Evolution and Pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China”(文章链接:http://www.sciencedirect.com/science/article/pii/S0966842X17301531)。
免疫研究所粟硕教授为论文第一作者,扬州大学兽医学院顾敏副研究员为论文共同第一作者,中国科学院微生物研究所的刘翟研究员和高福院士以及中国科学院武汉病毒所的崔杰研究员等为论文的主要作者。扬州大学兽医学院刘秀梵院士和动物医学院周继勇教授为论文通讯作者。据悉,细胞出版社出版的《Trends in Microbiology》创刊于1993年,年发文量100篇左右,该期刊2016年影响因子为11.02,近5年平均影响因子为10.239。
自2013年3月中国发现了首例人感染H7N9禽流感病毒病例以来,每年冬春季都会出现一波人感染的疫情,截止到2017年5月23日止已有五波共1486人感染H7N9流感病毒,死亡率约为38%。2016年10月开始的第五波H7N9流感病毒感染人的疫情中,不仅感染人数增加,而且在地理位置上出现了向我国西部省份扩散的现象。从第五波疫情开始,部分H7N9病毒出现了变异,变成了高致病性毒株,出现了高致病性H7N9流感病毒感染人的病例。同时在湖南、河北、河南、天津和陕西等省市的家禽养殖场也开始爆发新发的高致病性H7N9禽流感病毒疫情,超过30万羽鸡被扑杀。H7N9流感病毒不仅对人类健康造成巨大威胁,也严重威胁我国的养殖业。因此,新出现的高致病性H7N9禽流感疫情受到全社会的高度关注。
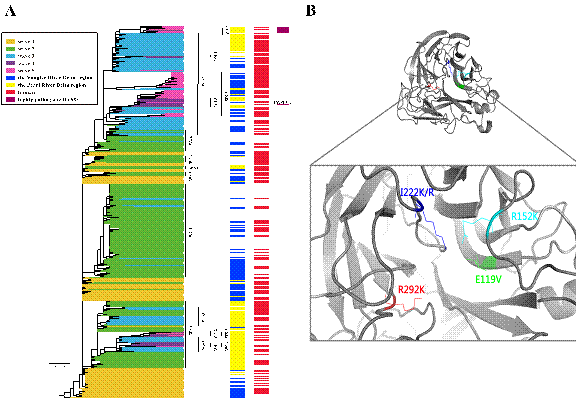
基于这些背景,由南京农业大学免疫研究所牵头,多个单位的研究人员对2013年以来的五波H7N9流感病毒疫情从病例临床特征、分布范围、病毒的进化趋势和生物学特征等方面进行了系统流行病学研究和分析。疫情特征表现为,珠三角与长三角地区病例数显著高于其他地区,男性病例显著高于女性病例。针对H7N9流感病毒目前在我国的流行特点和基因组序列进化特征,研究人员首次通过遗传进化大数据分析发现,2015-2016年流行的第四波H7N9流感疫情,病毒全部由之前的三波病毒进化而来,一部分来源于长三角和珠三角地区共同流行的clade Wave 3-2分支;另一部分则由全部来源于珠三角的clade Wave 3-1分支进化而来,并且全部分布于珠三角区域。2016年底出现的第五波H7N9流感疫情,主要由第四波在长三角和珠三角地区共同流行的clade Wave 3-2分支进化而来,其中最新出现的高致病性H7N9毒株均来自于珠三角区域并形成一个独立的分支,提示高致病性H7N9流感病毒起源于珠三角地区。通过氨基酸位点分析,发现新出现的H7N9流感病毒HA蛋白获得了高致病性禽流感病毒裂解位点的特征,NA蛋白出现了R292K、E119V、 R152K , I222K/R的突变,揭示了潜在的多重耐药特征,提示未来需加强临床上的耐药监测,以便及时调整治疗方案。
高致病性H7N9禽流感病毒的出现,不仅威胁人类健康,也给我国的养禽业带来巨大的经济损失,应持续加强对该病毒的监测。本研究首次通过对五波H7N9疫情中分离的H7N9病毒进行系统遗传进化分析揭示H7N9病毒在我国的遗传演化特征和流行特征,为防控新发的高致病性H7N9流感病毒感染人和高致病性H7N9在禽中的爆发提供重要指导和理论依据。
图1:(A)截止到2017年4月,H7N9流感病毒感染人的五波疫情中感染病例数与死亡数统计。(B)用RAxML方法系统展示了H7N9流感病毒HA基因的进化树,以病毒shanghai/1/2013 H7N9为根。高致病性H7N9流感病毒用深紫色表示。使用蓝色,黄色与没有颜色来区分病毒的分离于长三角地区,珠三角地区还是其他地区。红色表示人感染的H7N9病毒分离株,无色表示环境或者禽类的H7N9病毒分离株。
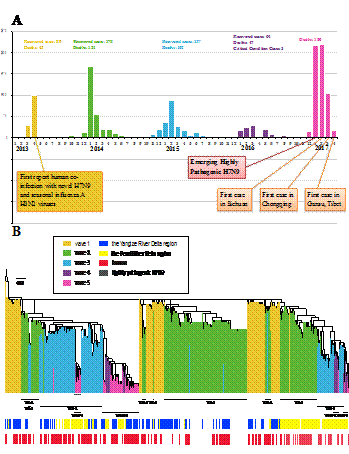
图2:(A)用RAxML方法系统展示了H7N9流感病毒NA基因的进化树。(B)利用3D结构模型解析了H7N9流感病毒NA蛋白上的耐药基因位点变化。